
Исследование
механизмов регуляции экспрессии генов
предполагает анализ статистических
особенностей контекстной организации
последовательностей ДНК и установление
закономерностей, связывающих эти
особенности с функцией гена и
молекулярными механизмами его регуляции.
Открытие закономерностей в регуляторных
последовательностях ДНК имеет две стороны:
(1) установление правил (закономерностей),
связывающих контекстную организацию и
функцию последовательности; (2)
установление контекстных свойств
промоторных районов и использование их для
функциональной аннотации генов (предсказание
класса промотора по нуклеотидной
последовательности). Оценивается как
точность итогового предсказания, так и
статистическая значимость правил,
оцененная по контрастной выборке данных.
Отличительная
особенность используемого алгоритма "Дискавери"
(Discovery – Открытие) – использование
специфических схем (паттернов)
характеристик, которые описывают лишь
подгруппы всего обучающего набора объектов
(обучающей выборки последовательностей).
Входные данные конвертируются в форму
высказываний логики первого порядка (присутствие
либо отсутствие сигналов в
последовательности). Дальнейший поиск
паттернов выполняется с помощью
вероятностных оценок (Kovalerchuk and Vityaev, 2000;
Витяев, 1992). Алгоритм "Дискавери"
генерирует гипотезы в виде
параметрического семейства формул типа:
A1&…&An
=>
A0 ,
где
A0,A1,…,An – логические
выражения (включающие логические связки AND,
OR, NOT, скобки и произвольные арифметические
выражения с параметрами).
Параметрами
могут быть номера признаков, интервалы
изменения признаков, выделенные значения
признаков, параметры, модифицирующие
признак (подвергающие его различным
преобразованиям) и т.д. Система позволяет
реализовать перебор гипотез с помощью
стратегии, представляющей собой
семантический вероятностный вывод.
Уточнения гипотез осуществляются путем
добавления новых условий в посылку, либо
применением подстановок. Было показано, что
система "Дискавери" способна
обнаружить закономерности в языке первого
порядка, имеющие максимальные оценки
условной вероятности (Витяев, 1993; Kovalerchuk and
Vityaev, 2000). Для решения этой задачи система
автоматически определяет по таблицам
характеристик статистически значимые
правила в логике первого порядка. Такие
таблицы контекстных характеристик для
проанализированных выборок были
предварительно подготовлены с помощью
программ построения контекстных и
конформационных характеристик
нуклеотидных последовательностей (Витяев
и др., 2001; Kolchanov et al., 2003).
Компьютерная
система "Gene Discovery" основана на
адаптации алгоритма "Discovery" для
анализа нуклеотидных последовательностей
регуляторных районов с использованием
вспомогательных программ подготовки
выборок данных нуклеотидных
последовательностей, расчета
статистических, конформационных и
структурных свойств ДНК в форме логических
и вещественнозначных характеристик,
визуального представления закономерностей
и интерфейса пользователя. Система
разрабатывалась автором совместно с
коллективом ученых и программистов из ИЦиГ
СО АН и ИМ СО РАН (Витяев и др., 2001). На защиту
выносятся положения, связанные с
применением системы для анализа промоторов
генов эукариот.
Система "Gene Discovery" включает в себя модуль поиска закономерностей в таблицах молекулярно-биологических баз данных, статистически значимо коррелированных с классом регуляторных последовательностей и модуль распознавания промоторов в нуклеотидной последовательности на основе отобранных закономерностей (Витяев и др., 2001). Принципиальная схема системы представлена на рисунке 2.6.
Рис. Блок-схема
системы "Gene Discovery". Данные обозначены
прямоугольниками, процессы обработки
информации – овалами (Витяев и др., 2001).
На
вход системы подается обучающая выборка
нуклеотидных последовательностей двух
альтернативных классов: класс 1 – промоторы;
класс 2 – последовательности, не
выполняющие этой функции (например,
случайные последовательности с теми же
частотами нуклеотидов, выборки экзонов,
интронов и т.д.).
Имеется
блок программ, осуществляющих поиск
контекстных сигналов в
последовательностях этих двух классов.
Сигнал может быть:
·
контекстным
(короткое олигонуклеотидное слово,
функциональный сайт и т.д.),
·
конформационным
(участок ДНК, характеризующийся
особенностями конформационных или физико-химических
свойств, например, легкоплавкие участки ДНК,
сильно изогнутая ДНК и т.д.),
·
структурным
(например, участок низкой сложности текста,
шпилька вторичной структуры РНК и др.).
Система
реализована на языке С++ в среде VisualStudio 6.0 и
предназначена для интерактивной работы на
персональном компьютере.
Компьютерная
программа может принимать на входе сигналы
различной природы – контекстные и
структурные, – размеченные как короткие
участки на анализируемой
последовательности. В качестве структурных
сигналов использовались участков низкой
сложности, выделенные с помощью программы
LowComplexity (Orlov and Potapov, 2004; http://wwwmgs2.bionet.nsc.ru:8080/low_complexity/),
разработанной автором. Также в качестве
дополнительных сигналов с помощью
программы VMM рассчитывался нуклеосомный
потенциал – как способность
последовательности к формированию
нуклеосомы, оцененная статистически по
сходству с обучающей выборкой нуклеосомных
сайтов (Orlov and Levitsky, 2004). В качестве
контекстных сигналов использовались
весовые матрицы сайтов связывания
транскрипционных факторов, определенные на
основе базы данных TRRD и базы данных SELEX (http://wwwmgs.bionet.nsc.ru/mgs/gnw/selex/).
Далее при анализе промоторных районов
будет рассмотрены контекстные сигналы
только одного типа – несовершенные
олигонуклеотиды. Для выделения
олигонуклеотидных сигналов, специфичных к
данной группе промоторов, использовалась
программа ARGO, разработанная О.В. Вишневским
(Babenko et al.,
1999; Vishnevsky et al., 2004;
http://wwwmgs.bionet.nsc.ru/mgs/programs/argo/). Под
олигонуклеотидным сигналом, или мотивом,
понимается слово, записанное в обобщённом
15-буквенном алфавите IUPAC (Nomenclature Committee of the
International Union of Biochemistry (NC-IUB), 1986): {A, T, G, C, R=G/A, Y=T/C,
M=A/C, K=T/G, W=A/T, S=G/C, B=T/C/G, V=A/G/C, H=A/T/C, D=A/T/G, N=A/T/G/C}.
Программа
ARGO анализирует две контрастных выборки
последовательностей, одна из которых
выполняет специфичную функцию (позитивная
выборка, в нашем случае – промоторы), а
вторая лишена этой способности (негативная
выборка). В результате анализа этих выборок
с помощью системы ARGO выявляются
олигонуклеотидные мотивы, удовлетворяющие
следующим условиям: (1) мотив с высокой
частотой встречается в
последовательностях позитивной выборки и с
низкой – в последовательностях выборки
негативной; (2) различия частот
встречаемости мотива в двух этих выборках
статистически достоверны. Использовались
мотивы длиной 8 оснований, что
соответствует в среднем размеру корового
района ССТФ.
Комплексные
сигналы ищутся в форме логических
закономерностей. Закономерности имеют
форму гипотез типа ЕСЛИ-ТО (IF-THEN). Например,
условие "IF ANANANCA = 1 AND GWAKAWAW = 1" означает,
что олигонуклеотиды ANANANCA и GWAKAWAW должны быть
представлены в анализируемой
последовательности. Заключение "THEN Class =
1" означает, что последовательность
принадлежит к классу 1 (классу промоторов).
Комплексные сигналы строятся как выражения S1& S2& S3…&Sk, где k>1. (В приведенном выше примере k=2). Программа автоматически определяет оптимальное число сигналов в комплексном сигнале (паттерне). Ограничения на взаимное расположение сигналов в нуклеотидных последовательностях могут быть жесткими, с учетом позиций, либо мягкими, учитывающими только относительные позиции. Пример поиска сигналов взаимного присутствия (без учета позиций) приведен на рисунке.
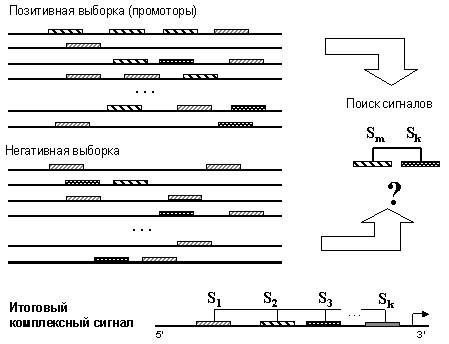
Рис. Задача поиска
комплексных сигналов в промоторах.
Олигонуклеотидный паттерн (комплексный
сигнал) S1&S2&S3…&Sk,
локализованный относительно старта
транскрипции (обозначенного стрелкой),
представлен в нижней части рисунка.
Показано схематическое распределение всех
олигонуклеотидов, используемых в качестве
характеристик при построении паттерна, в
последовательностях позитивной и
негативной выборок.
Комплексные
сигналы могут задаваться с учетом взаимной
локализации и ориентации олигонуклеотидов.
Рассмотрим простейший комплексный сигнал (S1,
S2), образованный парой
олигонуклеотидов, который задается
следующим образом:
(S1,
S2) = ( Pos(S1) < Pos(S2) & (Sign(S1)
= z1) & (Sign(S2) = z2) ) .
Здесь S1
и S2 – олигонуклеотиды в таблице "объект-признак";
Pos(S1) и Pos(S1) – позиции данных
олигонуклеотидов в некоторой
последовательности, Sign(S1) и Sign(S2)
– знак олигонуклеотида; z1,z2 Î
{+,-} знак означает, что олигонуклеотид
расположен в прямой (+) или комплементарной
(-) цепи.
Согласно
формуле (2.17) комплексный сигнал (S1, S2)
образован двумя сигналами S1 и S2 ,
расположенными так, что сигнал S1
находится левее сигнала S2 (Pos(S1)
< Pos(S2)) и, кроме того, каждый из
сигналов располагается в определенной
ориентации (Sign(S1)=z1)&(Sign(S2)=z2).
Гипотеза
о связи комплексного сигнала (S1, S2)
с классом промоторов записывается
следующим логическим выражением:
"R$S1,S2
( (S1, S2) =>
(Class(R) = l) ) ,
где R – некоторая нуклеотидная
последовательность; Class(R) – номер класса, к
которому принадлежит данная
последовательность (1 – промоторы, 2 –
случайные последовательности).
Гипотеза
утверждает, что любая последовательность R
относится к классу промоторов (классу l),
если в ней существуют сигналы S1 и S2,
расположенные так, что Pos(S1)<Pos(S2)
и такие, что их ориентации будут совпадать с
ориентациями z1 и z2.
Блок
поиска закономерностей "Gene Discovery"
перебирает в таблице "объект-признак"
все возможные варианты гипотезы для
комплексных сигналов (Si,Sj), где i,j=1,...N,
N – число индивидуальных сигналов (олигонуклеотидных
мотивов). Для каждого варианта по точному
критерию независимости Фишера для таблиц
сопряженности признаков оценивается связь
соответствующей комбинации с классом
объектов (например: класс 1 – промотор,
класс 2 – не-промотор), определяемая уровнем
значимости P(N1,N2,N3,N4)
критерия Фишера.
|
Класс 1 |
Класс 2 |
|
|
Условие
(1) выполнено |
N1 |
N2 |
|
Условие
(1) не выполнено |
N3 |
N4 |
|
Общее
число объектов в классе |
N1+N3 |
N2+N4 |
Здесь:
N1, N2 число промоторов (класс
1) и не-промоторов (класс 2), соответственно,
содержащих сигналы S1, S2,
удовлетворяющие условию; N3, N4
– число промоторов и не-промоторов,
соответственно, не удовлетворяющих
заданному условию.
Помимо
значения критерия Фишера оценивается также
условная вероятность PC(N1,N2)=
N1/(N1+N2)
отнесения последовательности к классу
промоторов при наличии в ней комплексного
сигнала (S1, S2). Эта вероятность
может рассматриваться как оценка точности
распознавания, обычно требуется, что эта
оценка не была менее 0.7.
При
анализе таблицы "Объект-признак"
система стартует с простейших парных
комплексных сигналов
(S1, S2) и
проводит последовательный направленный
поиск все более сложных сигналов,
постепенно усложняя их путем добавления
новых индивидуальных сигналов.
Следовательно, в общем случае под
комплексным сигналом мы понимаем
сигнал (S1,S2,...Sm) при m>1,
удовлетворяющий условию:
(Pos(S1) < Pos(S2))&(Pos(S2) < Pos(S3))&…&(Pos(Sm-1) < Pos(Sm))&(Sign(S1) = z1)& (Sign(S2) = z2)&…& (Sign(Sm) = zm) .
Проверяемая
гипотеза о связи комплексного сигнала (S1,S2,...Sm)
с классом промоторов в формальной логике
записывается следующим образом:
"R$S1,S2,...Sm
((S1,S2,...Sm) =>
Class(R) = l ) ,
где R – нуклеотидная
последовательность; Class(R) – номер класса, к
которому принадлежит данная
последовательность, m= 1,2,...– число
сигналов (олигонуклеотидов) в
рассматриваемой гипотезе.
Добавление
в комплексный сигнал новых индивидуальных
сигналов осуществляется так, чтобы
условная вероятность PC(N1,N2)
отнесения последовательности к классу
промоторов строго возрастала при условии,
что значение критерия Фишера P(N1,N2,N3,N4)
не выходит за пороговый уровень 0.05. В
результате подобного поиска формируется
полная группа комплексных сигналов Q = {(S11,...S1m1),
... ,(Si1,...Simi), ... ,(Sn1,...,Snmn)}
и соответствующих им промоторных
закономерностей, характеризующих
взаимосвязь комплексного сигнала с классом
последовательности. Таким образом, j-му
промотору сопоставляется подмножество
комплексных сигналов Qj Ì Q, находящихся в нем.
В
соответствии с задачами исследования были
проанализированы следующие типы данных:
(1)
короткие последовательности ДНК,
содержащие сайты связывания белковых
транскрипционных факторов, донорные и
акцепторные сайты сплайсинга.
(2)
протяженные последовательности ДНК
геномов эукариот, содержащие: (а) регуляторные
районы транскрипции, промоторы, энхансеры; (б)
5'-нетранслируемые последовательности
генов эукариот; (в) экзоны и интроны интрон-содержащих
генов эукариот; (г) сайты формирования
нуклеосом.
(3)
полные последовательности бактериальных
геномов (130 последовательностей).
(4)
полные последовательности хромосом ряда
геномов эукариот, включая все хромосомы
генома человека (релиз 34, сборка 2003 г.),
хромосомы дрожжей Saccharomyces cerevisiae и Schizosaccharomyces
pombe, хромосомы Arabidopsis thaliana и фрагменты
хромосом некоторых других организмов.
В
качестве источников информации
использовались база данных регуляторных
районов транскрипции эукариот TRRD (Kolchanov et
al., 2002), база данных промоторов эукариот EPD
(Perier et al., 2000; Praz et al., 2002), база данных сайтов
сплайсинга SpliceDB (Burset et
al., 2001), база данных экзонов и интронов
интрон-содержащих генов EID (Exon-Intron Database, Saxonov et
al., 2000), база данных нуклеотидных
последовательностей GenBank (Benson et
al., 2000; 2003). Для получения
последовательностей полных бактериальных
геномов и контигов хромосом человека
использовались информационные ресурсы
Национального Центра Биотехнологической
Информации США (National Center for Biotechnology Information –
NCBI, http://www.ncbi.nlm.nih.gov/), Европейского института
биоинформатики (EBI, http://www.ebi.ac.uk/) и
международный банка данных TAIR, содержащего
экспериментальные данные по модельному
растению Arabidopsis thaliana, (http://www.arabidopsis.org/,
Huala et al., 2001).
Протяженные
последовательности функциональных районов
геномной ДНК эукариот содержали
промоторные районы, фазированные
относительно старта транскрипции.
Последовательности были получены из базы
данных регуляторных районов транскрипции
TRRD (Kolchanov et al., 2002) и
базы данных промоторов эукариот EPD (Perier et al., 2000; Praz et al.,
2002). В базе данных TRRD представлено немного
меньше регуляторных последовательностей,
причем информация подробна качественно,
отмечены только экспериментально
установленные ССТФ, возможно разбиение на
функциональные группы промоторов, при этом
длина последовательностей промоторов в
зависимости от размера флангов исходно не
ограничена. Больший объем данных
представлен в БД EPD (Perier et
al., 2000; Praz et al., 2002).
Релиз EPD 76 за 2003
год содержит 2997 последовательностей длиной
600 нуклеотидов фазированных относительно
старта транскрипции как [-499;+100]. Однако
только 255 генов имеют разметку промотора,
картированную на геномную
последовательность из БД EMBL. Выборки
промоторов были извлечены из базы данных EPD,
в соответствии с принадлежностью к группам
организмов – промоторы генов позвоночных,
генов растений и генов прямокрылых.
Выборки
эукариотических промоторов были
составлены по БД TRRD следующим образом: по
принципу работы генов в одной генной сети –
гены липидного метаболизма, гены
эндокринной системы, по принципу
тканеспецифичности – гены эритропоэза (эритроид-специфичные),
гены регуляции холестерина, и по принципу
совместной индукции – глюкокортикоид-регулируемые
гены, интерферон-регулируемые гены, гены
ответа на тепловой шок. Выборки были
фазированы [-300;+100] относительно старта
транскрипции.
Выборки
экзонов и интронов извлекались из базы
данных EID (Exon-Intron Database) (Saxonov
et al., 2000; http://mcb.harvard.edu/gilbert/EID).
База данных сеодержала 25,130 белок-кодирующих
генов, содержащих интроны, для которых
разметка экзон-интронных границ была
подтверждена экспериментально.
Составлялись как выборки всех экзонов в
гене, так и выборки только первых, только
вторых, только третьих и т.д. экзонов.
Использовалась
выборка последовательностей ДНК,
содержащих экспериментально определенный
участок формирования нуклеосом (связывания
с гистоновым октамером) (Ioshikhes and Trifonov, 1993), –
всего 171 последовательность длиной 400 п.о.
Также исследовались выборки участков
формирования нуклеосом,
классифицированные по принадлежности
нуклеотидных последовательностей к
группам организмов (позвоночные, растения),
из баз данных "Samples" (http://wwwmgs.bionet.nsc.ru/cgi-bin/mgs/nsamples/)
и Nucleosome database (Levitsky et al.,
1999; 2004). Кроме того, использовались выборки
нуклеотидных последовательностей,
стабильность связи которых с гистоновым
октамером в составе нуклеосомы
определялась с помощью SELEX экспериментов ((Widlund
et al., 1997; Cao et al., 1998; Levitsky et al.,
2004).
Промоторы
содержат в своем составе сайты связывания
транскрипционных факторов (ССТФ),
встречаемость и расположение которых
определяют особенности экспрессии генов.
Ранее было показано (Kondrakhin et
al., 1995), что наиболее насыщен сайтами
связывания участок [-100;+1] относительно
старта транскрипции, поэтому исследовались
промоторные районы, содержащие именно этот
участок и фланкирующие его районы.
Один из
методов исследования контекстной
организации промоторов – поиск
неслучайных олигонуклеотидных слов (с
возможными несовпадениями). Такие
контекстные сигналы могут быть сайтами
связывания транскрипционных факторов (в
том числе – еще неизвестными). Промоторы
содержат также короткие участки ДНК с
характерными свойствами нуклеотидного
состава и физико-химическими параметрами –
конформационные сигналы. Такие участки
могут характеризоваться, например,
повышенной гибкостью двойной спирали ДНК,
отличаться температурой плавления и т.д.
Конформационные сигналы также могут быть
найдены статистически.
Ставилась
задача поиска контекстных закономерностей
промоторов, по которым можно определить,
содержит ли анализируемая
последовательность промотор, локализовать
старт транскрипции, предсказать класс
транскрибируемого гена и тип его регуляции.
Были рассмотрены промоторы генов системы
эритропоэза, эндокрин- и интерферон-регулируемых
генов, генов ответа на тепловой шок, генов
системы регуляции холестерина и генов
клеточного цикла, извлеченные из базы
данных TRRD. Было выполнено множественное
выравнивание выборок последовательностей
промоторов с помощью пакета Clustal,
установлено отсутствие протяженных
гомологичных участков.
Далее
контекстные сигналы искались в форме
специфичных вырожденных олигонуклеотидов.
Были проанализированы контрастные выборки
последовательностей, одна из которых
выполняет специфичную функцию (позитивная
выборка, в нашем случае
– промоторы), а вторая лишена этой
способности (негативная выборка
– случайные последовательности). В
результате анализа этих выборок с помощью
системы ARGO (Babenko et al.,
1999; Vishnevsky et al., 2004;
http://wwwmgs.bionet.nsc.ru/mgs/programs/argo/) выявлены
олигонуклеотидные мотивы, удовлетворяющие
следующим условиям: (1) мотив с высокой
частотой встречается в
последовательностях позитивной выборки и с
низкой – в
последовательностях выборки негативной; (2)
различия частот встречаемости любого
мотива в двух этих выборках статистически
достоверны (Vishnevsky et al.,
2004).
Последовательности
промоторов имели длину 300 п.о. (от –200 до +100 п.о.
относительно старта транскрипции). Именно
такие участки промоторов обладают
достаточной консервативностью при
множественном выравнивании и оценках
сходства. С помощью программ парного
выравнивания были удалены
последовательности с высоким уровнем
гомологии. Для любой пары из оставшейся
выборки сходство не превышало 70%.
Негативная выборка "не-промоторов"
содержала 1000 случайных
последовательностей той же длины и с теми
же частотами нуклеотидов, что и
последовательности промоторов.
При
анализе промоторов генов эндокринной
системы с помощью программы ARGO было
выявлено 68 специфичных олигонуклеотидов в
15-буквенном коде (далее они также будут
называться мотивами). Примеры приведены в
таблице 3.2 (Витяев и др., 2001).
Вторая
колонка таблицы 3.2 содержит описание
специфичных олигонуклеотидных мотивов
длины 8 в 15-буквенном коде, третья и
четвертая – долю последовательностей
позитивной и негативной выборок, в которых
данный мотив встретился хотя бы один раз.
Так, например, мотив KNCMAGDG встретился в 32
процентах выборки промоторов и лишь в 3
процентах случайных последовательностей.
Таблица
Примеры
олигонуклеотидных мотивов, специфичных для
промоторов генов эндокринной системы (в
районе –100, +20 относительно старта
транскрипции)
|
№пп |
Запись в 15-буквенном
коде |
Доля
последовательностей обучающей выборки,
в которых присутствует данный мотив* |
|
|
Промоторы |
Не-промоторы |
||
|
1 2 3 4 5 6 7 8 9 10 ... 68 |
KNCMAGDG KRCCWGNR ANANANCA GKNCAGRG CANAGCMN RGSNRGRG KGRSSAGR KGRSCNGR YRGRGNCA CWGWGNCN ... CTGNNCAN |
0.325000 0.350000 0.275000 0.225000 0.250000 0.400000 0.275000 0.375000 0.250000 0.300000 ... 0.250000 |
0.031000 0.044000 0.032000 0.010000 0.013000 0.041000 0.017000 0.043000 0.028000 0.042000 ... 0.035000 |
Примечание
к таблице
* – Различия между
промоторами и случайными
последовательностями статистически
достоверны в соответствии с биномиальным
распределением нуклеотидов. Пропущенные
данные отмечены многоточием.
Аналогичным
образом была проанализирована выборка,
содержащая 41 промоторную
последовательность эритроид-специфичных
генов, взятых из базы данных EpoDB (Stoeckert et
al., 1999) и других выборок промоторов,
построенных по базе данных TRRD.
Итак, с
помощью программы ARGO были найдены короткие
олигонуклеотидные мотивы, характерные для
рассматриваемых групп промоторов. На
следующем этапе осуществлялся поиск
локализации каждого из выявленных мотивов
в каждой из последовательностей
анализируемой группы промоторов.
Результаты поиска представлялись в виде
таблицы "объект–признак".
Таблица
Таблица
"объект-признак", описывающая
олигонуклеотидные сигналы, выявленные в
промоторах генов эндокринной системы
|
Номер
последовательности в выборке* |
Номер
контекстного сигнала |
Последовательность
контекстного сигнала |
Позиция
начала сайта относительно старта
транскрипции |
Ориентация** |
Класс*** |
|
1 |
6 |
RGSNRGRG |
-65 |
1 |
1 |
|
1 |
6 |
RGSNRGRG |
-60 |
1 |
1 |
|
1 |
8 |
KGRSCNGR |
-100 |
-1 |
1 |
|
1 |
66 |
YTSCWGNW |
+13 |
-1 |
1 |
|
1 |
67 |
TCMAGNMN |
+13 |
1 |
1 |
|
... |
... |
... |
... |
... |
... |
|
40 |
67 |
TCMAGNMN |
-65 |
-1 |
1 |
|
40 |
68 |
CTGNNCAN |
-79 |
1 |
1 |
|
40 |
68 |
CTGNNCAN |
-61 |
1 |
1 |
|
41 |
1 |
KNCMAGDG |
-51 |
1 |
2 |
|
41 |
10 |
CWGWGNCN |
-13 |
-1 |
2 |
|
... |
... |
... |
... |
... |
... |
|
1040 |
56 |
HNNKGCTG |
-64 |
1 |
2 |
|
1040 |
56 |
HNNKGCTG |
-12 |
1 |
2 |
|
1040 |
64 |
NCWGGGNC |
-8 |
1 |
2 |
Примечания к таблице:
* – В таблице с номера 1 по 40 представлены промоторы, с номера 41 по 1041 – случайные последовательности с тем же нуклеотидным составом. Из-за большого объема (16300 строк) данные не могут быть приведены полностью. Пропущенные строки отмечены многоточием.
** – Ориентация: 1-
прямая цепь, -1 – комплементарная цепь.
*** – Класс
последовательности 1 – промоторы, 2 – не-промоторы.
Сигналами регуляции экспрессии генов могут служить не только сайты связывания транскрипционных факторов (ССТФ), но и участки ДНК с аномальными свойствами, такими как низкая сложность текста, повторы определенного типа, участки вториччной структуры ДНК и РНК (для 5'-нетраслируемых районов генов). Предсказание и разметка таких участков выполнялась с помощью авторских программ, разработанных участниками проекта.
Была согласована разметка (формат) для метаописания данных, используемая в системах GeneDiscovery и ExpertDiscovery.
Пример приведен ниже.
<motifs>
Signal_Number 266
<Signal 1>
name GGRRNSRG
method_name Weight_Matrics
Parameter_Number 0
</Signal 1>
<Signal 2>
name NKGGGANG
method_name Weight_Matrics
Parameter_Number 0
</Signal 2>
<Signal 3>
name YCTCHSYH
method_name Weight_Matrics
Parameter_Number 0
</Signal 3>